Stochastic processes code¶
Gillespie Algorithm¶
- By solving the master equation the time evolution of the probability distribution can be calculated.
- Unfortunately, the master equation is very difficult to solve, either numerically or analytically.
- In most cases, we therefore do not aim to solve the master equation. Rather a trajectory of individual transitions that is consistent with the master equation is simulated.
The most well known algorithm was proposed by D. Gillespie:
- The system is in \(x\) at time \(t\)
- Estimate probabilities \(w_i\) for all feasible transitions from this state \(x → x´\)
- Estimate the time \(\Delta t\) until which the transition happens
- Estimate which transition happens. The probability for an individual transition is proportional to \(w_i\).
- Update the state \(x\) and time \(t\): \(t → t + \Delta t\)
"""
Stochastic simulation of gene expression using Gillespie.
"""
import numpy as np
import pandas as pd
from matplotlib import pyplot as plt
def stochastic(tend: float=100, omega: float=1, b: float=5.0, info: bool=True) -> pd.DataFrame:
"""
The parameters are b, dp, dm and the mean number of proteins A0.
omega is a system size parameter (volume). In the case of omega=1, the concentration
corresponds to the number of molecules
:param tend: end time of simulation
:param omega: system size parameter
:Param b: burst size, average number of proteins per mRNA
:return: stochastic gillespie timecourse
"""
# parameter
A0 = 100.0 # steady state value of protein
dm = 2.0
dp = 0.2
kp = b * dm
km = A0 * dp/b
M0 = km/dm # steady state value of mRNA
if info:
print("[M0]={}; [A0]={}".format(M0, A0))
# initial conditions
M = 0
A = 0
# simulation
t = 0 # [min]
ix = 0
res = []
while t<tend:
res.append([t, M/omega, A/omega])
ix = ix + 1
# calculate the rates for all transitions
w1 = omega * km # transcription (M,A) -> (M+1, A)
w2 = dm * M # decay mRNA (M,A) -> (M-1, A)
w3 = kp * M # translation (M,A) -> (M, A+1)
w4 = dp * A # decay protein (M,A) -> (M, A-1)
rate = w1 + w2 + w3 + w4
# estimate time
eps1 = 1-np.random.random() # np.random: Uniformly distributed floats over [0, 1)
dt = -np.log(eps1)/rate
t = t + dt
# which transition was selected
rate_rand = rate * np.random.random()
if rate_rand < w1:
M = M + 1 # transcription
elif rate_rand < w1 + w2:
M = M - 1 # decay mRNA
elif rate_rand < w1 + w2 + w3:
A = A + 1 # translation
elif rate_rand < w1 + w2 + w3 + w4:
A = A - 1 # decay protein
return pd.DataFrame(res, columns=["time", "M", "A"])
def plot_results(dfs, omega, b, **kwargs):
""" Helper function for plotting.
:param df:
:return:
"""
fig, (ax1, ax2) = plt.subplots(nrows=1, ncols=2, figsize=(10,5))
ax1.set_title("M (mRNA), omega={}, b={}".format(omega, b))
ax1.set_ylabel("# mRNA")
ax2.set_title("A (protein), omega={}, b={}".format(omega, b))
ax2.set_ylabel("# protein")
if not isinstance(dfs, list):
dfs = [dfs]
for df in dfs:
ax1.plot(df.time, df.M, color="blue", **kwargs)
ax2.plot(df.time, df.A, color="red", **kwargs)
for ax in (ax1, ax2):
ax.set_xlabel("time [min]")
plt.show()
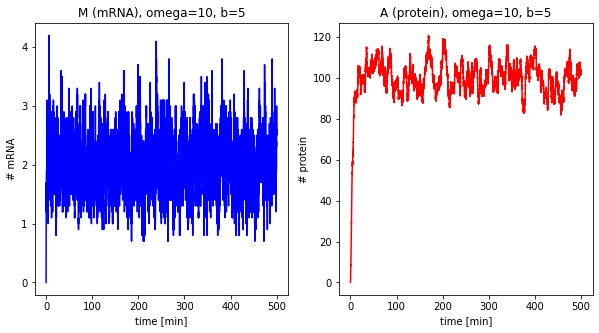
Single trajectories¶
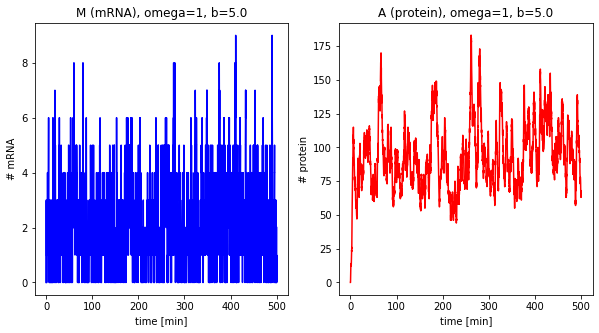
df = stochastic(tend=500, omega=1, b=5.0)
plot_results(df, omega=1, b=5.0)
[M0]=2.0; [A0]=100.0
Effect of system size¶
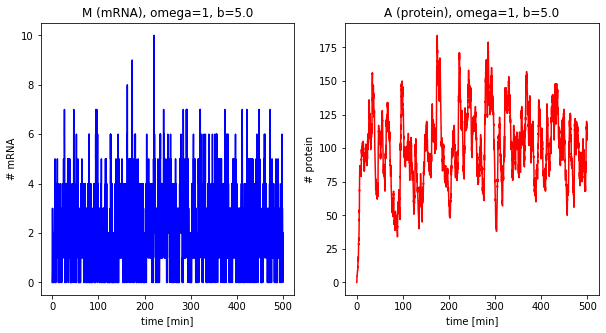
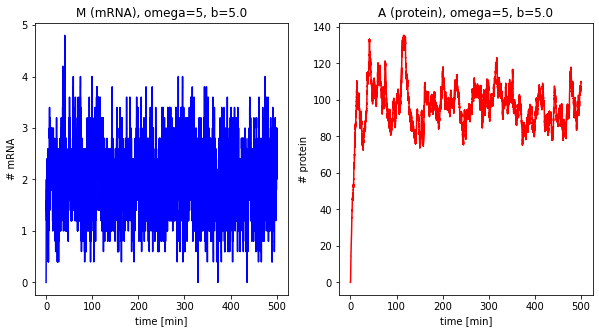
Now we change the system size parameter \(\Omega\).
dfs_omega = []
for omega in [1, 5, 10, 25]:
df = stochastic(500, omega=omega, b=5.0)
plot_results(df, omega=omega, b=5.0)
dfs_omega.append(df)
[M0]=2.0; [A0]=100.0
[M0]=2.0; [A0]=100.0
[M0]=2.0; [A0]=100.0
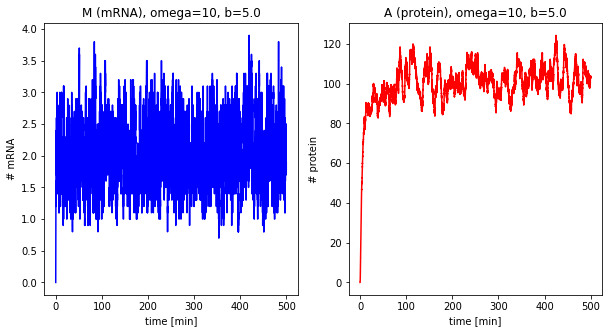
[M0]=2.0; [A0]=100.0
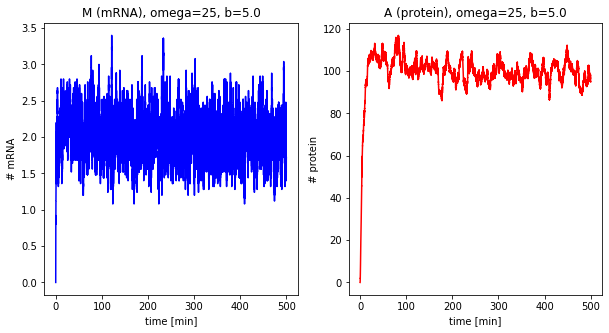
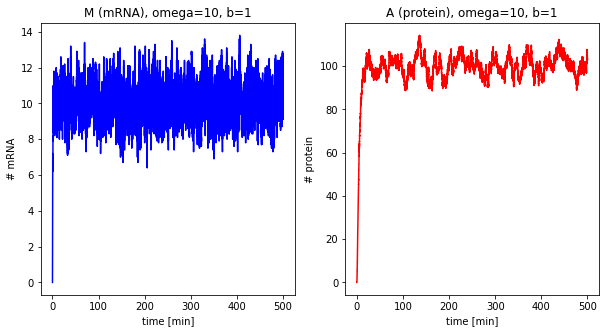
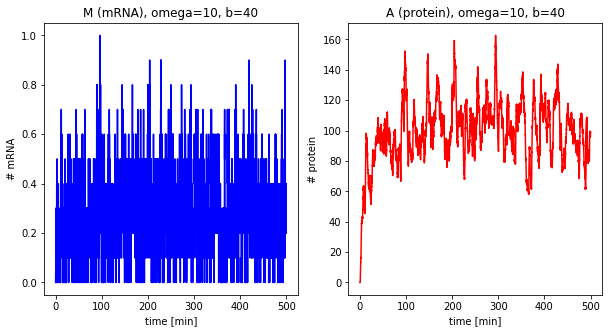
Effect of the burst size¶
The next step is looking at the burst size of the system, i.e., how many proteins are translated per mRNA.
df_b = []
for b in [1, 5, 40]:
df = stochastic(500, omega=10, b=b)
plot_results(df, omega=10, b=b)
[M0]=10.0; [A0]=100.0
[M0]=2.0; [A0]=100.0
[M0]=0.25; [A0]=100.0
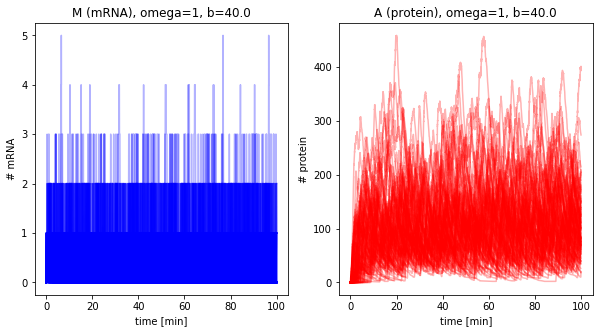
Sampling trajectories from master equation¶
omega = 1
b = 40.0
dfs = []
for k in range(100):
df = stochastic(100, omega=omega, b=b, info=False)
dfs.append(df)
plot_results(dfs, omega=omega, b=b, alpha=0.3)
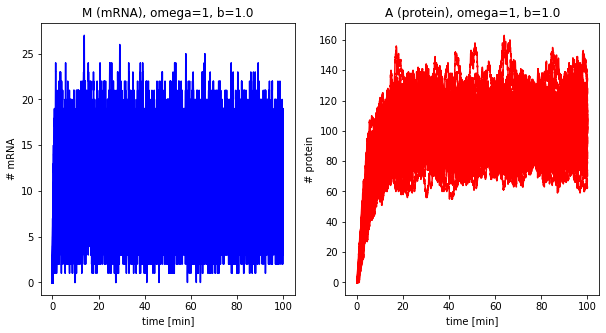
omega = 1
b = 1.0
dfs = []
for k in range(100):
df = stochastic(100, omega=omega, b=b, info=False)
dfs.append(df)
plot_results(dfs, omega=omega, b=b)